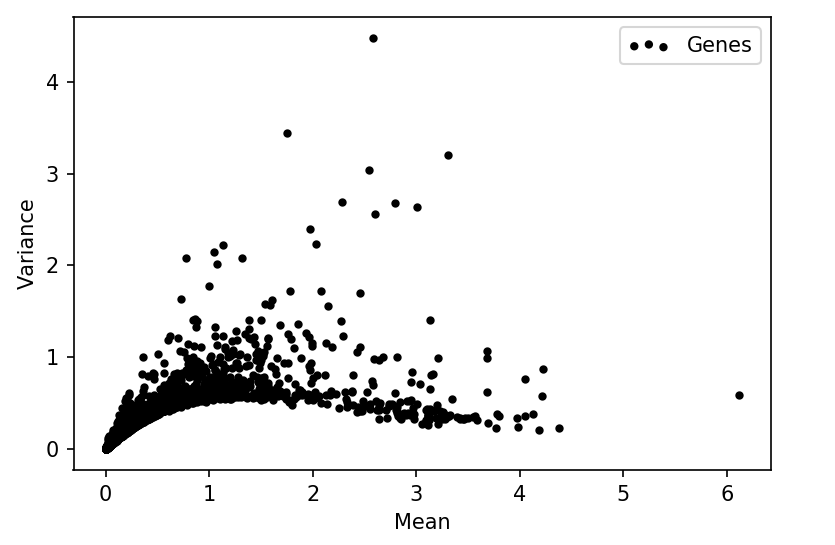
Converting images of line graphs to data
Recently Google released aggregated anonymized data on community mobility for 131 countries based on cell phone location data. The data spanned the time period between February 16 and March 29, and made clear how people have limited visits to places categorized as “Retail & recreation”.
The data was made available as line graphs in PDF reports per country, meaning you need to open each country you want to look at in a separate document. I wanted to see how population behaviour had changed globally by looking at many countries at the same time. But the data used to make the curves in the reports was not available for direct download.
In the PDF’s the curves are vector graphics, so in theory the underlying data is embedded within the PDF. Using Illustrator I could access the curve element, but I couldn’t see a way to extract the data that builds the curve. I tried finding tools explicitly made for extracting data from PDF’s and encountered a tool called “norma”, but I couldn’t get it to work.
The line graphs are pretty clear and have simple colors, so I thought it would be pretty simple to extract the values even if I had the bitmap images of the graphs.
I used Greenshot to take screenshots of the regions in the reports in the range -80% to 80%, which are the labeled extremes on the y-axis, while covering the entire x-axis.

After loading a screenshot in Python I could isolate the pixels belonging to the curve by using a threshold on the red channel.

These pixel values together with the knowledge of the axis scales can be used to recover the original data.
# Binarize the image
mask = 0.5 < img[:, :, 0]
# The `.argmin(0)` method will return the first index where
# we see a black pixel along the y-axis in the image.
# Images have negative y-axis, flip to get positive values.
pixel_value = -mask.argmin(0)
# We know the y-axis spans the interval [-80, 80]. To get the
# original values, scale values to the fraction of vertical pixels
# the observed values are at. Then scale these fractions to the
# [-80, 80] interval.
percent = 160 * (pixel_value + img.shape[0]) / img.shape[0] - 80
# The reported dates start on February 16 and end on March 29.
# Make a DateRangeIndex defined for a single day's resolution.
dates = pd.date_range(datetime.date(2020, 2, 16), datetime.date(2020, 3, 29))
daily_percent = (
pd.DataFrame({
# Use the `cut()` function to bin horizontal pixels into
# intervals corresponding to the days.
'bins': pd.cut(np.arange(percent.shape[0]), bins=dates.shape[0]),
'percent': percent
})
# Group the percent values by the day bins.
.groupby('bins')
# Extract the median percent value for each bin.
.median()
.reset_index(drop=True)
)
# Finally, add the dates to the data
daily_percent['date'] = datesNow we have the percentage values for each day in a DataFrame and can re-plot it to recreate the reported curve.
p.options.figure_size = (6, 4)
(
p.ggplot(p.aes(x='date', y='percent'), daily_percent)
+ p.geom_line()
+ p.scale_y_continuous(limits=(-80, 80))
+ p.theme_minimal()
)
Having validated that it was possible to recover the data given the screenshots, I spent some time creating screenshots for 47 countries. With these I could simply loop over the images, create the DataFrames, and combine them at the end. With the combined data I was able to make the combined plot I wanted:

The notebook I used to do this, as well as a CSV of the resulting DataFrame, are available on Github.